




Blog

Pharmacovigilance en Afrique et au moyen orient
La pharmacovigilance, une science ayant pour but de détecter, évaluer et prévenir les effets indésirables des médicaments, elle est essentielle pour la santé publique. En Afrique et moyen orient, un développement des règlementations a vu le jour ces dernières années et décennies, avec une intégration croissante au niveau des réseaux régionaux et internationaux.
-
Concernant les pays du Maghreb et du moyen orient :
En Tunisie, le Centre National de Pharmacovigilance (CNPV) a été créé sous la tutelle du ministère de la Santé Publique, depuis 1984 par la Loi 84-84. Récemment la Tunisie a développé une règlementation plus détaillée et des directives des Bonnes Pratiques de Pharmacovigilance ».
Au Maroc, le centre Antipoison et de Pharmacovigilance (CAPM) a été créé en 1989.
En Algérie, le Centre National de Pharmacovigilance et de Matériovigilance (CNPM) a été créé en 1998.
En Égypte, c’est le centre de pharmacovigilance «â€¯The Egyptian Pharmacovigilance Center » (EPVC) qui la gère.
En Irak Le Centre de Pharmacovigilance Irakien (IPhvC) a été créé en 2010 sous le ministère de la Santé.
En Jordanie, le centre de pharmacovigilance Jordanien «â€¯The Jordanian Pharmacovigilance Center » (JPC) travaille en collaboration avec l’administration des aliments et des médicaments (FDA) depuis 2001.
En 2003, le gouvernement de l’Arabie Saoudite établis la SFDA et en 2009 une unité séparée appelé le Centre National de Pharmacovigilance (NPC) a été créé.
Seuls 45 % des pays arabes sont membres officiels du Centre de collaboration pour la surveillance internationale des médicaments de l'Organisation mondiale de la santé (OMS). Des pays comme le Maroc, la Tunisie, l'Arabie saoudite, l'Égypte et la Jordanie sont considérés comme des pays avancés en matière de pharmacovigilance, tandis que d'autres pays n'en sont qu'aux premiers stades de la mise en œuvre et du développement de la pharmacovigilance pour l'application de la majorité des activités liées au système de santé, y compris la pharmacovigilance.
Vu la diversité des systèmes de santé et le développement des centres de pharmacovigilances dans le Maghreb et le moyen orient, un guide s’est avéré essentiel pour l’uniformité et l’harmonisation des pratiques et des réglementations de pharmacovigilance entre les pays arabes.
Ainsi la ligne directrice sur les bonnes pratiques de pharmacovigilance pour les pays Arabe (Guideline on good Pharmacovigilance practices (GVP) For Arab Countries) a été développé en étant largement inspiré des bonnes pratiques européennes de pharmacovigilance (EU GVP) et des ICH, il est considéré comme «â€¯modèle idéale » à adopter pour les pays arabes qui peuvent ajouter d’autres spécificités locales.
-
Concernant d’autres pays de l’Afrique :
En Afrique centrale, la République Démocratique de Congo, le Système National de Pharmacovigilance est fonctionnel depuis 2009 année de la création du Centre National de Pharmacovigilance (CNPV), qui depuis 2010, est un membre effectif du centre international de pharmacovigilance de l’OMS l’Uppsala Monitoring Centre (UMC).
En Afrique de l’Ouest Au Burkina Faso, depuis 2008, création d’un service national de pharmacovigilance au sein de la Direction générale de la pharmacie, du médicament et des laboratoires (DGPML).
En Côte d’Ivoire, la pharmacovigilance est soutenue par la AIRP (Autorité Ivoirienne de Régulation Pharmaceutique) et un centre de pharmacovigilance est en cours de formation.
Le Centre de Pharmacovigilance au Sénégal est sous l’égide de l’Agence sénégalaise de Réglementation pharmaceutique (ARP).
Le Centre National de Pharmacovigilance au Nigeria a été créé dans le cadre du Programme International de Surveillance des Médicaments de l’Organisation mondiale de la santé (OMS) en 2004.
Le Centre National de Pharmacovigilance au Ghana est géré par l’Autorité des Aliments et Médicaments (FDA). Le Ghana a rejoint le Programme de Surveillance Internationale des Médicaments de l’OMS en novembre 2001
Au Mali, c’est le Centre National de Référence de Pharmacovigilance (CNRP) crée au sein du centre national d’appui à la lutte contre la maladie (CNAM) en 2011, qui est chargé de cette activité.
En Togo, la première activité formelle de pharmacovigilance a été notée en décembre 2006.
En Afrique de l’Est, en Ouganda, sous l’autorité national des médicaments (National Drug Authority (NDA), le centre national de pharmacovigilance d’Ouganda a été créé en 1993. En 2020, l’autorité national des médicaments en Ouganda, est en voie de création d’une application mobile appelée «â€¯Med Safety », afin de faciliter la collecte d’informations.
En Afrique du Sud, la pharmacovigilance est une fonction du Conseil de contrôle des médicaments (the Medicines Control Council (MCC)) et ce depuis 1997.
Plusieurs autres pays ont initié la mise en place et l’organisation de systèmes nationaux de pharmacovigilance. Toutefois, d’autres actions et moyens s’avèrent nécessaires afin de généraliser un suivi de pharmacovigilance efficace dans tous les pays de l’Afrique.

Medical devices regulation in Africa
Medical devices play a crucial role in the public health protection. It was therefore necessary to frame them with an adequate legal arsenal in order to guarantee access to safe, high-quality products.
- What is a Medical Device?
According to the Global Harmonisation Task Force (GHTF), medical devices (MDs) are defined as any article, instrument, device or machine (including medical software and applications) intended for use, alone or in combination, for medical purposes and its action is not obtained by pharmacological, immunological, or metabolic means.”
In vitro diagnostic medical devices (IVMD) are a subset of MDs intended for in vitro examination of samples from the human body to provide information for diagnostic, control, or compatibility purposes. They include reagents, calibration agents, control materials and test kits.
- Nomenclature and classification
The establishment of a MD nomenclature would support efforts to strengthen the assessment, the regulation the management and the access to medical devices. The most widely used classifications worldwide are the World Nomenclature of Medical Devices (WNMD), the Universal System of Nomenclature of Medical Devices (USNMD), the United Nations Standard Classification of Products and Services (UNSCPS) and the European Nomenclature of Medical Devices (ENMD).
According to the WHO, MDs are classified according to their level of risk to the patient. The level of risk is defined according to criteria such as the duration of use by the patient, the invasivity (invasive or not, degree of invasion), the possibility or not of reuse, the therapeutic or diagnostic aim, the dependence of an energy source (active or not).
There are different classifications such as classification (I, IIa, IIb, and III) by EMDN and classification (A, B, C, and D) by GHTF.
- Global MD regulation:
The majority of industrialized MD-producing countries (Australia, Canada, Japan, USA and European Union countries) have a regulatory system covering the entire life cycle of a MD since the assessment of conformity to quality requirements, the safety and clinical efficacy of a product until the marketing authorization and surveillance of medical devices (vigilance).
MD regulations are country-specific; however, most countries do not have a regulatory framework for importing and marketing medical devices. For this purpose, the IMDRF (International Medical Device Regulators Forum) undertakes to serve as a basis for the development of national regulations. To learn about national policies for MDs, the World Atlas of Medical Devices published in 2022 provides an overview of regulatory frameworks in each WHO member state around the world.
At European level, there is a developed legislative and a reference framework providing: definitions, fields of application, classification, general conditions for marketing and commissioning of MDs, essential health and safety requirements for MDs, conformity certification procedures (CE). Regulation (EU) 2017/745 and Regulation (EU) 2017/746 set the rules for the marketing of MD and DMDIV and the rules applicable to related clinical investigations.
- MD regulation in Africa
Most Member States with medical device regulatory authorities are in the WHO African Region such as: Angola, Benin, Botswana, Burkina Faso, Cameroon, Ivory Coast, Ethiopia, Eritrea, Gabon, Gambia, Ghana, Guinea, Kenya, Mauritania, Mozambique, Namibia, Nigeria, Uganda, Democratic Republic of Congo, United Republic of Tanzania, Rwanda, Senegal and Sierra Leone.
The West African Economic and Monetary Union (WAEMU) commission has strengthened the regulatory framework for MD of its Member States by drawing up guidelines on classification, marketing authorization, the statement and communication of MDs and the safety and performance requirements of DMs. This guide has been well adopted and implemented by Ivory Coast, Senegal, and Burkina Faso. However, some African countries, such as Ivory Coast, are making progress in implementing their national regulations for MD.
In 2021, the African Medical Device Regulators Forum (AMDRF), with support from WHO, implemented a harmonized regulatory framework for the regulation of medical devices, including in vitro diagnostics (IVDs). Guidelines have been developed on the regulatory requirements for the deliverance of a market authorization for medical devices, including in vitro diagnostic medical devices, registration of medical device establishments, guidelines for the import and export of medical devices, including in vitro diagnostic medical devices, and guidelines for the inspection of manufacturing sites for the assessment of the medical device quality management system based on ISO 13485:2016. The development of regional guidelines promotes regulatory harmonization, accelerates regulatory decision-making processes, and ensures timely access to these important health products for users.
.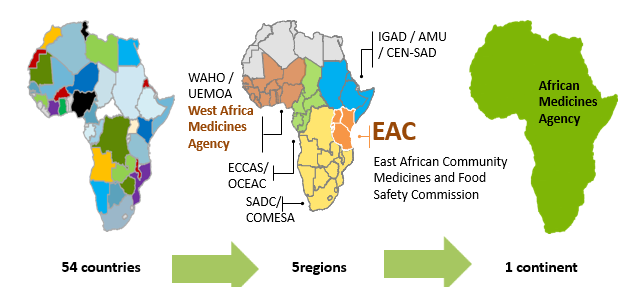
The regulatory situation in Africa: a complex landscape
In recent decades, there have been several processes to harmonise pharmaceutical regulations around the world. Africa has joined the same dynamic and progress in forming different pharmaceutical regulatory groups to harmonize as much as possible the regulations throughout its different regions and strengthen its capacities facing public health problems.
In this context, the Regional Committee of the World Health Organization (WHO) has adopted for Africa, a regional strategy on medical product regulation to improve the governance of medical product regulatory systems in its Member States; strengthen the capacity of national pharmaceutical regulatory authorities to perform regulatory functions and strengthen regional regulatory harmonisation and convergence.
Several national and regional initiatives have been performed to strengthen and support the regional harmonisation of standards and regulatory policies in the African continent. The African Union (AU) comprises 8 regions and has 55 member states: Central Africa (9 states), East Africa (14 states), North Africa (7 states), Southern Africa (10 states) and West Africa (15 states).
- National Regulatory Systems in Africa:
At national level, all Member States have set up regulatory systems with varying functional capacities and various levels of maturity from one country to another.
The number of countries that have autonomous or semi-autonomous regulatory bodies reached 51% of countries in 2020. These member states are Algeria, Benin, Botswana, Burkina Faso, Comoros, Ivory Coast, Eritrea, Ethiopia, Gambia, Ghana, Kenya, Liberia, Madagascar, Malawi, Namibia, Nigeria, Uganda, Democratic Republic of Congo, United Republic of Tanzania, Rwanda, Sierra Leone, South Sudan, and Zambia.
To date, four African national regulatory authorities (NRAs), namely Tanzania, Ghana, Egypt, and Nigeria, have reached maturity three for pharmaceuticals products, a designation attributed by the World Health Organization (WHO) to a country with a stable, functional, and integrated regulatory system. Egypt has reached Maturity Level 3 in the regulation of locally produced and imported vaccines, while the others have reached this level for medicines and imported vaccines.
- The African Medicines Regulatory Harmonization (AMRH) Initiative
At regional level, the Harmonisation of Medicines Regulations in Africa (HMRA) initiative has been implemented in the different regional economic communities (RECs) officially recognized by the African Union.
This initiative has permitted to formulate proposals for the harmonization of pharmaceutical regulations within the different regional economic communities, namely: the Southern African Development Community (SADC) in Southern Africa, the Economic and Monetary Community of Central Africa (CEMAC) and the Economic Community of Central African States (ECCAS) in Central Africa, the East African Community (EAC) in East Africa as well as the Economic Community of West African States (ECOWAS) and the West African Economic and Monetary Union (WAEMU) in West Africa and the Intergovernmental Authority for Development (IGAD).
Although all RECs implement programmes to harmonise drug regulation, various levels of maturity have been achieved. Indeed, the EAC, the SADC through the collaboration ZANZIBONA, ECOWAS and WAEMU have succeeded to put guidelines for the registration and authorization of pharmaceutical products.
- The African Medicines Agency (AMA)
In order to support and promote the regional efforts within the framework of AMRH, the African Medicines Agency (AMA) was established in January 2015 and officially came into effect on November 5, 2021. A total of 23 Member States have signed, ratified, and deposited the instrument of ratification: Algeria, Benin, Burkina Faso, Cameroon, Chad, Egypt, Gabo, Ghana, Guinea, Lesotho, Mali, Morocco, Mauritius, Namibia, Niger, Uganda, Rwanda, Saharawi, Seychelles, Sierra Leone, Senegal, Tunisia, and Zimbabwe.
Its main objective is to improve the capacity of Member States and RECs to regulate medical products in order to improve access to quality, safe and effective medical products on the African continent. It should also promote the adoption and harmonisation of policies and standards for the regulation of medical products, provide the necessary scientific guidance and coordinate existing regulatory harmonization efforts in RECs and Regional Health Organisations (RHOs) recognized by the African Union.
.
Les études de stabilité des médicaments : Etude de la situation en Afrique
I. Cadre général :
- La stabilité des médicaments : Facteur déterminant de la qualité
La stabilité d’un produit pharmaceutique est définie selon l’ICH, comme l’aptitude d’un médicament à conserver ses propriétés physiques, chimiques, microbiologiques et biopharmaceutiques dans des limites spécifiées pendant sa durée de validité.
- Les études de stabilité des médicaments :
Avant sa commercialisation et en vue d’octroi d’une AMM, un médicament est soumis à des essaies de stabilité dans des conditions standardisées et internationales. Cette stabilité doit être assurée pendant toute la durée de validité du médicament. Des lignes directrices ont été recommandées par l’ICH et l’OMS, afin de tester les variations de la qualité d’un PA ou d’un médicament au cours du temps en fonction de différents facteurs environnementaux (Température, Humidité, Lumière). Ces études permettront à l’industriel pharmaceutique de définir les conditions de conservation, de déterminer la durée de validité de ses produits, et de garantir aux patients des produits pharmaceutiques de qualité, d’efficacité et de sécurité.
II. Les lignes directrices des études de stabilité ICH et OMS
- Le guide ICH Q1A-Q1F
Le guide ICH des études de stabilité englobe un ensemble de 6 guides (Q1A-Q1F) détaillant, pour chacun d’eux, des aspects plus spécifiques. Le principal guide est le Q1A (R2), détaillant les études de stabilité à réaliser, lors de la mise sur le marché d’un nouveau PA ou d’une nouvelle spécialité, dans l’un des 3 régions ICH (Europe, Etats-Unis et Japon). Les principales exigences recommandées par le guide ICH sont la sélection des lots, le type d’emballage des échantillons, la fréquence des essais et les conditions générales de stockage (Température et Humidité relative) à tester.
- Les lignes directrices de l’OMS :
L’OMS rédige également son propre guide « Stability testing of active pharmaceutical ingredients and finished pharmaceutical product » (2009). Les lignes directrices recommandées par l’OMS sont quasiment identiques à celles de l’ICH à l’exception de quelques différences telles que la sélection des lots et des conditions générales de stockage.
La plus importante partie de ce guide est l’annexe 1 qui résume pour chaque pays membre de l’OMS les conditions à appliquer lors des études de stabilité à long terme.
En effet, l’OMS a défini 5 zones climatiques :
- Zone I (climat tempéré ; 21°C/45% RH),
- Zone II (Climat Subtropicale et Méditerranéen ; 25°C/60% RH),
- Zone III (Climat chaud et sec ; 30°C/35% RH),
- Zone IVA (Climat chaud et humide ; 30°C/65% RH),
- Zone IVB (Climat chaud et très humide ; 30°C/75% RH).
A chaque zone correspond les conditions à appliquer lors des études de stabilité à long terme. Chaque pays membre est donc classé dans l’une de ces quatre zones en fonction de deux critères : la température annuelle moyenne mesurée à l’air libre et la moyenne Exigences des études de stabilité en Afrique :
En Afrique, prenons l’exemple de la Tunisie, le Maroc, l’Algérie et les 8 pays de l’UEMOA (Bénin ; Burkina Faso ; Côte d’Ivoire ; Guinée-Bissau ; Mali ; Niger ; Sénégal et Togo), ces pays appliquent les lignes directrices ICH et OMS, dans les études de stabilité associées à l’enregistrement et la commercialisation des produits pharmaceutiques. Les pays du Maghreb (Tunisie, Maroc, Algérie) appartenant à la zone II de l’OMS, conduisent leurs études de stabilité à long terme à 25 °C/60% RH, alors que les pays membres de l’UEMOA, à l’exception du Togo, appartenant à la zone IVA appliquent 30 °C/65±5% RH. Le Togo appartient à la zone IVB et conduit ses essais de stabilité à 30 °C/75% RH.
Nous remarquons que les pays de l’Afrique du nord appliquent la même zone et exigence en humidité et température que pour la plupart des pays européens et que les autres pays africains appliquent des zones où la température et l'humidité sont supérieures ; ceci s’explique par les conditions climatiques différentes.
Il est aussi primordial pour l’enregistrement d’un produit importé dans ces pays, de finaliser l’étude de stabilité couvrant la durée de validité proposée.
AREMA vous propose grâce à son équipe d’experts et de partenaires locaux ainsi que sa plateforme REMAREG d’intelligence réglementaire, des études approfondies de la conformité de vos dossiers aux exigences de stabilité et autres exigences pour chaque pays désigné.
.
Résumé Pays: CAMEROUN
Le Cameroun est un pays de l’Afrique centrale ayant comme langue officielle le français. Le Cameroun importe près de 90% de ses besoins en médicaments et le marché est estimé à plus de 100 milliards de francs Cfa.
Le Cameroun fait partit de la Communauté économique et monétaire de l'Afrique centrale (CEMAC),
Les médicaments et autres produits de santé (tel que les dispositifs médicaux) ne peuvent être importés qu’après enregistrement.
Le dossier d’enregistrement des médicaments doit être en format CTD et en langue Française obligatoirement pour certaines parties. Le dossier d’enregistrement des dispositifs médicaux est spécifique au pays.
L’autorité régissant l’enregistrement : Direction de la Pharmacie et du Médicament et des laboratoires (DPML) , site web : https://dpml.cm/index.php/fr/
L’enregistrement des médicaments dépend de l’acceptation du dépôt au niveau de la DPML et du niveau de conformité du dossier et nécessite 12 mois à 24 mois.
AREMA vous propose une assistance totale pour l’enregistrement de vos médicaments au Cameroun. Pour plus d’informations : info@arema-international.com
.
Résumé pays: Tunisie
La Tunisie est un pays de l’Afrique du nord. Le marché des médicaments en Tunisie représente près de 1 milliards de dollar, avec environ 47% d’importations (sous le monopole de l’état à travers la Pharmacie centrale de Tunisie) et 53% comme production locale.
Les standards d’enregistrement des médicaments se rapprochent des standards européens avec un guide national d’enregistrement des médicaments.
Le dossier d’enregistrement doit être en format CTD et en langue Française ou anglaise avec certaines parties obligatoirement en Français et/ou Arabe.
L’autorité régissant l’enregistrement des médicaments : la DPM (Direction de la Pharmacie et du Médicament) dont le site web : http://www.dpm.tn/
L’enregistrement des médicaments en Tunisie nécessite une prise de rendez-vous , dès acceptation du dépôt l’évaluation est effectuée par la DPM et par le LNCM (Laboratoire National de contrôle des médicaments) et nécessite 2 à 3 ans pour enregistrement du médicament en Tunisie.
Les dispositifs médicaux, les compléments alimentaires et les cosmétiques suivent une autre procédure spécifique d’AMC (autorisation de mise à la consommation)
AREMA vous propose une assistance totale , pour plus d’informations : info@arema-international.com
.
Résumé Pays: Sénégal
Le Sénégal est un pays de l’Afrique de l'Ouest dont la capitale est Dakar ayant une population d’environ 15 millions d’habitants, et comme langue officielle le français.
Le Sénégal est un état membre de la Communauté économique des États de l'Afrique de l'Ouest (CEDEAO).
Les médicaments et autres produits de santé ne peuvent être importés qu’après enregistrement.
L’autorisation de mise sur le marché est valide pour une période de 5 années.
Enregistrement des Médicaments au Sénégal :
Le Sénégal est membres d’institutions régionales dont principalement l’UEMOA et applique donc ses directives concernant l’enregistrement des médicaments. De plus il y a certains textes nationaux et certaines particularités du pays à connaitre.
Le dossier d’enregistrement doit être en format CTD et en langue Française.
L’autorité régissant l’enregistrement des médicaments : ARP Senegal (Authorité de Regulation Pharmaceutique)
L’enregistrement des médicaments dépend de l’acceptation du dépôt au niveau de la DPM et du niveau de conformité du dossier et nécessite 9 mois à 24 mois.
AREMA vous propose une assistance totale pour vos médicaments et produits de santé au Sénégal.
.
Résumé Pays: Côte d'Ivoire
La Côte d’Ivoire, Abidjan est un pays de l’Afrique de l'Ouest ayant une population d’environ 27 millions d’habitant, et comme langue officielle le français.
La Côte d’Ivoire est un état membre de la Communauté économique des États de l'Afrique de l'Ouest (CEDEAO).
Les médicaments et autres produits de santé ne peuvent être importés qu’après enregistrement.
Une inspection du site de fabrication des médicaments est nécessaire lors des premiers enregistrements.
Marché des médicaments en Côte d’Ivoire
Le marché Africain en général est un marché à fort volume d’importation, en Afrique la production locale ne représente que 3 % de la production mondiale, alors que le marché est en pleine expansion et la demande toujours plus grande. La Côte d’Ivoire importe près de 94% de ses médicaments.
L’introduction des maladies non transmissibles s’ajoutent aux grandes pandémies que connaît le continent font que la demande de médicaments n’a jamais été aussi forte.
Arema vous offre la possibilité d’effectuer une étude de marché et des enquêtes terrain selon votre besoin.
Enregistrement des Médicaments
La Côte d’Ivoire est membres d’institutions régionales dont principalement l’UEMOA et applique donc ses directives concernant l’enregistrement des médicaments. De plus il y a certains textes nationaux et particularité du pays à connaitre.
Le dossier d’enregistrement doit être en format CTD et en langue Française.
L’autorité régissant l’enregistrement des médicaments est la DPML (Direction de la Pharmacie, du médicament et les laboratoires , dont le site web : www.dpml.ci/
L’enregistrement des médicaments dépend du niveau de conformité du dossier et des dates des commissions, il nécessite au départ de prendre un RDV pour le dépôt qui dépendra de la disponibilité des autorités. Lla délivrance de l’Autorisation de mise sur le marché prendra 12 mois à 2 ans après le dépôt.
AREMA vous propose une assistance totale pour l’enregistrement de vos médicaments en Côte d’Ivoire.
Nous fournissons les services réglementaires depuis la veille réglementaire, la préparation et traduction de vos dossiers, le dépôt, le suivi et l’enregistrement de vos différents produits jusqu’à la mise en place d’un système de pharmacovigilance et de suivi post-AMM.
.
Résumé Pays : Le Burkina Fasso, Principales informations marché et réglementaires
-
Le Burkina Fasso est un pays de l’Afrique de l'Ouest dont la capitale est Ouagadougou ayant une population d’environ 20 millions d’habitant, et comme langue officielle le français.
Le Burkina Fasso est un état membre de la Communauté économique des États de l'Afrique de l'Ouest (CEDEAO).
Les médicaments ne peuvent être importés au Burkina Fasso qu’après enregistrement.
Marché des médicaments au Burkina Fasso:
Le marché Africain en général est un marché à fort volume d’importation, en Afrique la production locale ne représente que 3 % de la production mondiale, alors que le marché est en pleine expansion et la demande toujours plus grande. Le Burkina Fasso possède une seule unité de fabrication.
De plus l’introduction des maladies non transmissibles s’ajoutent aux grandes pandémies que connaît le continent font que la demande de médicaments n’a jamais été aussi forte.
Enregistrement des médicaments :
Le Burkina Faso est membres d’institutions régionales dont principalement l’UEMOA et applique donc ses directives concernant l’enregistrement des médicaments. De plus il y a certains textes nationaux et particularités du pays à connaitre.
Le dossier d’enregistrement doit être en format CTD et en langue Française.
L’autorité régissant l’enregistrement des médicaments : Agence Nationale de régulation pharmaceutique du Burkina Faso (ANRP) https://anrp.bf
-
L’enregistrement des médicaments dépend du niveau de conformité du dossier et nécessite 9 mois à 18 mois.
AREMA vous propose une assistance totale pour l’enregistrement de vos médicaments au Burkina Fasso.
Nous fournissons les services réglementaires depuis la veille réglementaire, la préparation et traduction de vos dossiers, le dépôt, le suivi et l’enregistrement de vos différents produits jusqu’à la mise en place d’un système de pharmacovigilance et de suivi post-AMM.
Afin d’initier toute collaboration ou pour toute consultation vous pouvez nous contacter par email:
-

New formulation of Carbetocin could save thousands of women’s lives
According to a study published in June 27, 2018, led by WHO in collaboration with MSD for Mothers and Ferring Pharmaceuticals. A new drug formulation of Carbetocin could save thousands of women’s lives
Approximately 70 000 women die every year because of post-partum haemorrhage – increasing the risk that their babies also die within one month.
Currently, WHO recommends oxytocin as the first-choice drug for preventing excessive bleeding after childbirth. Oxytocin, however, must be stored and transported at 2–8 degrees Celsius, which is hard to do, in many countries, depriving many women of access to this lifesaving drug. When they can obtain it, the drug may be less effective because of heat exposure.
The study, published in the New England Journal of Medicine, has shown an alternative drug – heat-stable carbetocin – to be as safe and effective as oxytocin in preventing postpartum haemorrhage. This new formulation of carbetocin does not require refrigeration and retains its efficacy for at least 3 years stored at 30 degrees celsius and 75% relative humidity.
For more information here is the article: https://www.nejm.org/doi/full/10.1056/NEJMoa1805489
and WHO press release : http://www.who.int/news-room/detail/27-06-2018-who-study-shows-drug-could-save-thousands-of-women%E2%80%99s-lives
.
CPP use for Drugs Registration in Africa and Middle east
The Certificate of Pharmaceutical Product (CPP) is a document of the World Health Organization’s (WHO) Certification Scheme on the quality of pharmaceutical products moving in international commerce. CPP also known as CoPP establishes the status of the pharmaceutical product and of the applicant for this certificate in the exporting country.WHO developed CPP in the late 1960s with the aim to provide assurance to its members about the quality of the pharmaceutical products moving in international trade.The CPP is widely required by emerging countries such as African and middle eastern countries in new drugs’ submission processes, post-approval changes and renewal of drugs’ registrations. Some countries in middle east request its legalization by their embassy in country of origin. CPP is more and more replacing marketing authorization in country of origin in these countries and is accurately checked by health authorities as a reference document for drug approval process.For more information : http://www.who.int/medicines/areas/quality_safety/regulation_legislation/certification/guidelines/en/index1.html
.