




Blog

Pharmacovigilance in Africa and the Middle East
Pharmacovigilance, the science of detecting, assessing and preventing adverse drug reactions, is essential to public health. In Africa and the Middle East, regulations have developed over recent years and decades, with increasing integration into regional and international networks.
-
Concerning the Maghreb and Middle East countries:
In Tunisia, the National Pharmacovigilance Centre (CNPV) was created under the supervision of the Ministry of Public Health in 1984 by Law 84-84. Recently, Tunisia has developed more detailed regulations and guidelines for Good Pharmacovigilance Practice.
In Morocco, the Anti-Poison and Pharmacovigilance Center (CAPM) was created in 1989.
In Algeria, the National Centre for Pharmacovigilance and Materiovigilance (CNPM) was created in 1998.
In Egypt, pharmacovigilance is managed by the Egyptian Pharmacovigilance Center (EPVC).
In Iraq, the Iraqi Pharmacovigilance Center (IPhvC) was created in 2010 under the Ministry of Health.
In Jordan, the Jordanian Pharmacovigilance Center (JPC) has been working in collaboration with the Food and Drug Administration (FDA) since 2001.
In 2003, the government of Saudi Arabia established the SFDA and in 2009 a separate unit called the National Pharmacovigilance Center (NPC) was established.
Only 45% of Arab countries are official members of the World Health Organization's (WHO) Collaborating Centre for International Medicines Surveillance.
Countries such as Morocco, Tunisia, Saudi Arabia, Egypt and Jordan are considered advanced in terms of pharmacovigilance, while other countries are only in the early stages of implementing and developing pharmacovigilance for the majority of health system activities, including pharmacovigilance.
Given the diversity of health systems and the development of pharmacovigilance centres in the Maghreb and the Middle East, a guide has proven to be essential for the uniformity and harmonization of pharmacovigilance practices and regulations between Arab countries.
The Guideline on good Pharmacovigilance practices (GVP) For Arab Countries has been developed on the basis of the European Good Pharmacovigilance Practices (EU GVP) and the ICH, and is considered an "ideal model" for Arab countries, which can add other local specificities.
-
Concerning other African countries:
In Central Africa, the Democratic Republic of Congo, the National Pharmacovigilance System has been operational since 2009, when the National Pharmacovigilance Centre (CNPV) was created. Since 2010, the CNPV has been an effective member of the WHO's international pharmacovigilance center, the Uppsala Monitoring Centre (UMC).
In West Africa in Burkina Faso, since 2008, a national pharmacovigilance service has been created within the Directorate General of Pharmacy, Medicines and Laboratories (DGPML).
In Côte d'Ivoire, pharmacovigilance is supported by the AIRP (Ivorian Pharmaceutical Regulatory Authority), and a pharmacovigilance centre is being formed.
The Pharmacovigilance Centre in Senegal is under the aegis of the Senegalese Pharmaceutical Regulatory Agency (ARP).
The National Pharmacovigilance Centre in Nigeria was established under the World Health Organization's (WHO) International Drug Surveillance Programme in 2004.
The National Pharmacovigilance Centre in Ghana is managed by the Food and Drugs Authority (FDA). Ghana joined the WHO's International Drug Monitoring Programme in November 2001.
In Mali, the National Reference Centre for Pharmacovigilance (CNRP), created within the National Centre for Disease Control Support (CNAM) in 2011, is responsible for this activity.
In Togo, the first formal pharmacovigilance activity was noted in December 2006.
In East Africa, Uganda, under the National Drug Authority (NDA), the Uganda National Pharmacovigilance Centre was established in 1993. In 2020, the National Drug Authority in Uganda is in the process of creating a mobile application called "Med Safety", in order to facilitate the collection of information.
In South Africa, pharmacovigilance has been a function of the Medicines Control Council (MCC) since 1997.
Several other countries have initiated the establishment and organization of national pharmacovigilance systems. However, other actions and resources are necessary in order to generalize effective pharmacovigilance monitoring in all African countries.

Medical devices regulation in Africa
Medical devices play a crucial role in the public health protection. It was therefore necessary to frame them with an adequate legal arsenal in order to guarantee access to safe, high-quality products.
- What is a Medical Device?
According to the Global Harmonisation Task Force (GHTF), medical devices (MDs) are defined as any article, instrument, device or machine (including medical software and applications) intended for use, alone or in combination, for medical purposes and its action is not obtained by pharmacological, immunological, or metabolic means.”
In vitro diagnostic medical devices (IVMD) are a subset of MDs intended for in vitro examination of samples from the human body to provide information for diagnostic, control, or compatibility purposes. They include reagents, calibration agents, control materials and test kits.
- Nomenclature and classification
The establishment of a MD nomenclature would support efforts to strengthen the assessment, the regulation the management and the access to medical devices. The most widely used classifications worldwide are the World Nomenclature of Medical Devices (WNMD), the Universal System of Nomenclature of Medical Devices (USNMD), the United Nations Standard Classification of Products and Services (UNSCPS) and the European Nomenclature of Medical Devices (ENMD).
According to the WHO, MDs are classified according to their level of risk to the patient. The level of risk is defined according to criteria such as the duration of use by the patient, the invasivity (invasive or not, degree of invasion), the possibility or not of reuse, the therapeutic or diagnostic aim, the dependence of an energy source (active or not).
There are different classifications such as classification (I, IIa, IIb, and III) by EMDN and classification (A, B, C, and D) by GHTF.
- Global MD regulation:
The majority of industrialized MD-producing countries (Australia, Canada, Japan, USA and European Union countries) have a regulatory system covering the entire life cycle of a MD since the assessment of conformity to quality requirements, the safety and clinical efficacy of a product until the marketing authorization and surveillance of medical devices (vigilance).
MD regulations are country-specific; however, most countries do not have a regulatory framework for importing and marketing medical devices. For this purpose, the IMDRF (International Medical Device Regulators Forum) undertakes to serve as a basis for the development of national regulations. To learn about national policies for MDs, the World Atlas of Medical Devices published in 2022 provides an overview of regulatory frameworks in each WHO member state around the world.
At European level, there is a developed legislative and a reference framework providing: definitions, fields of application, classification, general conditions for marketing and commissioning of MDs, essential health and safety requirements for MDs, conformity certification procedures (CE). Regulation (EU) 2017/745 and Regulation (EU) 2017/746 set the rules for the marketing of MD and DMDIV and the rules applicable to related clinical investigations.
- MD regulation in Africa
Most Member States with medical device regulatory authorities are in the WHO African Region such as: Angola, Benin, Botswana, Burkina Faso, Cameroon, Ivory Coast, Ethiopia, Eritrea, Gabon, Gambia, Ghana, Guinea, Kenya, Mauritania, Mozambique, Namibia, Nigeria, Uganda, Democratic Republic of Congo, United Republic of Tanzania, Rwanda, Senegal and Sierra Leone.
The West African Economic and Monetary Union (WAEMU) commission has strengthened the regulatory framework for MD of its Member States by drawing up guidelines on classification, marketing authorization, the statement and communication of MDs and the safety and performance requirements of DMs. This guide has been well adopted and implemented by Ivory Coast, Senegal, and Burkina Faso. However, some African countries, such as Ivory Coast, are making progress in implementing their national regulations for MD.
In 2021, the African Medical Device Regulators Forum (AMDRF), with support from WHO, implemented a harmonized regulatory framework for the regulation of medical devices, including in vitro diagnostics (IVDs). Guidelines have been developed on the regulatory requirements for the deliverance of a market authorization for medical devices, including in vitro diagnostic medical devices, registration of medical device establishments, guidelines for the import and export of medical devices, including in vitro diagnostic medical devices, and guidelines for the inspection of manufacturing sites for the assessment of the medical device quality management system based on ISO 13485:2016. The development of regional guidelines promotes regulatory harmonization, accelerates regulatory decision-making processes, and ensures timely access to these important health products for users.
.
The regulatory situation in Africa: a complex landscape
In recent decades, there have been several processes to harmonise pharmaceutical regulations around the world. Africa has joined the same dynamic and progress in forming different pharmaceutical regulatory groups to harmonize as much as possible the regulations throughout its different regions and strengthen its capacities facing public health problems.
In this context, the Regional Committee of the World Health Organization (WHO) has adopted for Africa, a regional strategy on medical product regulation to improve the governance of medical product regulatory systems in its Member States; strengthen the capacity of national pharmaceutical regulatory authorities to perform regulatory functions and strengthen regional regulatory harmonisation and convergence.
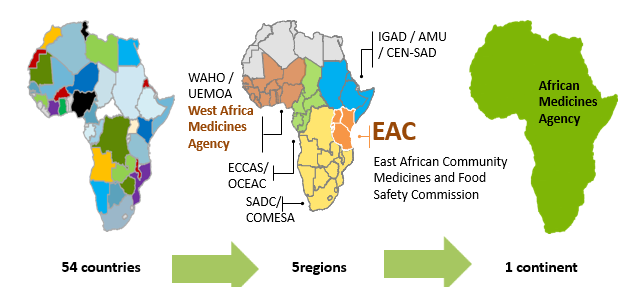
Several national and regional initiatives have been performed to strengthen and support the regional harmonisation of standards and regulatory policies in the African continent. The African Union (AU) comprises 8 regions and has 55 member states: Central Africa (9 states), East Africa (14 states), North Africa (7 states), Southern Africa (10 states) and West Africa (15 states).
- National Regulatory Systems in Africa:
At national level, all Member States have set up regulatory systems with varying functional capacities and various levels of maturity from one country to another.
The number of countries that have autonomous or semi-autonomous regulatory bodies reached 51% of countries in 2020. These member states are Algeria, Benin, Botswana, Burkina Faso, Comoros, Ivory Coast, Eritrea, Ethiopia, Gambia, Ghana, Kenya, Liberia, Madagascar, Malawi, Namibia, Nigeria, Uganda, Democratic Republic of Congo, United Republic of Tanzania, Rwanda, Sierra Leone, South Sudan, and Zambia.
To date, four African national regulatory authorities (NRAs), namely Tanzania, Ghana, Egypt, and Nigeria, have reached maturity three for pharmaceuticals products, a designation attributed by the World Health Organization (WHO) to a country with a stable, functional, and integrated regulatory system. Egypt has reached Maturity Level 3 in the regulation of locally produced and imported vaccines, while the others have reached this level for medicines and imported vaccines.
- The African Medicines Regulatory Harmonization (AMRH) Initiative
At regional level, the Harmonisation of Medicines Regulations in Africa (HMRA) initiative has been implemented in the different regional economic communities (RECs) officially recognized by the African Union.
This initiative has permitted to formulate proposals for the harmonization of pharmaceutical regulations within the different regional economic communities, namely: the Southern African Development Community (SADC) in Southern Africa, the Economic and Monetary Community of Central Africa (CEMAC) and the Economic Community of Central African States (ECCAS) in Central Africa, the East African Community (EAC) in East Africa as well as the Economic Community of West African States (ECOWAS) and the West African Economic and Monetary Union (WAEMU) in West Africa and the Intergovernmental Authority for Development (IGAD).
Although all RECs implement programmes to harmonise drug regulation, various levels of maturity have been achieved. Indeed, the EAC, the SADC through the collaboration ZANZIBONA, ECOWAS and WAEMU have succeeded to put guidelines for the registration and authorization of pharmaceutical products.
- The African Medicines Agency (AMA)
In order to support and promote the regional efforts within the framework of AMRH, the African Medicines Agency (AMA) was established in January 2015 and officially came into effect on November 5, 2021. A total of 23 Member States have signed, ratified, and deposited the instrument of ratification: Algeria, Benin, Burkina Faso, Cameroon, Chad, Egypt, Gabo, Ghana, Guinea, Lesotho, Mali, Morocco, Mauritius, Namibia, Niger, Uganda, Rwanda, Saharawi, Seychelles, Sierra Leone, Senegal, Tunisia, and Zimbabwe.
Its main objective is to improve the capacity of Member States and RECs to regulate medical products in order to improve access to quality, safe and effective medical products on the African continent. It should also promote the adoption and harmonisation of policies and standards for the regulation of medical products, provide the necessary scientific guidance and coordinate existing regulatory harmonization efforts in RECs and Regional Health Organisations (RHOs) recognized by the African Union.
.
Stability studies of pharmaceutical products : The African situation
I. General Framework :
- Stability of pharmaceutical products: Critical factor of quality
The stability of a pharmaceutical product is defined according to the ICH, as " the ability of a drug to maintain its physical, chemical, microbiological and biopharmaceutical properties within specified limits during its shelf life."
- Stability studies of pharmaceutical products:
Before being marketed and in order to grant the Marketing Authorization, a pharmaceutical product is subjected to stability assays under standardized and international conditions. This stability must be ensured throughout the validity period of the medicinal product. Guidelines have been recommended by the ICH and WHO, in order to test the variations of the quality of an active substance AP or a drug over the time, according to different environmental factors (Temperature, Humidity, Light). These studies will allow the pharmaceutical industry to define the storage conditions, to determine the period of validity of its products, and to guarantee patients pharmaceutical products of quality, efficacy, and safety.
II. The ICH et OMS guidelines
- The guideline ICH Q1A-Q1F
The ICH guideline of stability studies includes a set of 6 guidelines (Q1A-Q1F) detailing for each of them, more specific aspects. The main guideline is the Q1A (R2), detailing the stability studies to be conducted, when a new PA or a new specialty is subjected to the Marketing Authorization grant, in one of the tripartite regions of ICH (Europe, United States United and Japan). The main requirements recommended by the ICH guideline are the selection of batches, the type of sample packaging, the frequency of tests and the general storage conditions (temperature and relative humidity) to be evaluated.
- The WHO guidelines:
The WHO has drafted its own guideline « Stability testing of active pharmaceutical ingredients and finished pharmaceutical product » (2009). The recommended guidelines of WHO are almost identical to those of the ICH guidelines, except for a few differences such as batch selection and general storage conditions.
Annexe 1 of this guideline is the most important part, it summarizes for each WHO member country, the conditions to be applied during the long-term stability studies. Indeed, the WHO has defined 5 climatic zones:
- Zone I (Temperate climate; 21°C/45% RH),
- Zone II (Subtropical and Mediterranean Climate; 25°C/60% RH),
- Zone III (Hot and dry climate; 30°C/35% RH)
- Zone IVA (Hot and humid climate; 30°C/65% RH)
- Zone IVB (Hot and very humid climate; 30°C/75% RH).
Each zone corresponds to the conditions to be applied during long-term stability studies. Each member country is therefore classified in one of these zones according to two criteria: the average annual temperature measured in the open air and the annual average partial water vapor pressure.
II. Stability studies requirements in Africa:
In Africa, as examples Tunisia, Morocco, Algeria and the 8 member states of UEMOA (Benin; Burkina-Faso; Ivory Coast; Guinea-Bissau; Mali; Niger; Senegal et Togo), these countries apply ICH and WHO guidelines in stability studies associated with the registration and the marketing of pharmaceutical products. The Maghreb countries (Tunisia, Morocco, Algeria), belonging to WHO zone II, conduct their long-term stability studies at 25°C/60% RH, while UEMOA member states, except for Togo, belonging to the zone IVA apply 30°C/65±5% RH. Togo belongs to zone IVB and conducts its stability tests at 30°C/75% RH.
We note that the countries of North of Africa apply the same zone and requirement in terms of humidity and temperature as for the most European countries whereas, the other African countries apply zones where the temperature and humidity are higher; this is due to the different climatic conditions.
It is also essential for the registration of an imported product into these countries, to finalize the stability study during the proposed period of validity.
AREMA, own its experts’ team and local partners as well as its REMAREG regulatory intelligence platform, offers in-depth studies of the compliance of your documents with the stability requirements and other requirements of each of the designated country.
.
Country overview: CAMEROON
Cameroon is a central African country having French as official language. Cameroon imports nearly 90% of its needs in medicines. Knowing that the market is estimated at more than 100 billion CFA francs.
Cameroon is part of the Central African Economic and Monetary Community (CEMAC).
Medicines and other health products (such as medical devices) can be imported only after registration.
The registration dossier must be in CTD format and must be in French for some parts. Registration dossier for medical devices is specific to the country.
The authority governing the registration of medicines: Directorate of Pharmacy and Medicines and Laboratories (DPML), website: https://dpml.cm/index.php/en/
The registration of drugs depends on the acceptance of the deposit at the level of the DPML and the level of compliance of the file and requires 12 months to 24 months.
AREMA offers you total assistance for the registration of your medicines in Cameroon.
.
Country overview: Tunisia
Tunisia is a north African country. The drug market in Tunisia represents nearly 1 billion dollars, with about 47% of imports (under the monopoly of the government through Central Pharmacy of Tunisia) and 53% as local production.
Tunisia's drug registration standards are approaching European standards with a national drug registration guideline.
The registration file must be in CTD format and in French or English language with some parts in French and / or Arabic.
The authority governing the registration of medicines in Tunisia are: the DPM (Directorate of Pharmacy and Medicine) whose website: http://www.dpm.tn/
Medicine registration in Tunisia requires an appointment through a local and legal representative, upon acceptance of the deposit the assessment is made by the DPM and the LNCM (National Laboratory of Drug Control). It requires 2 to 3 years for registration of the drug in Tunisia.
Medical devices , food supplements and cosmetics are under another specific procedure of AMC (Autorization for consumption).
AREMA offers you full support. For more information : info@arema-international.com
.
Country overview: Senegal
Senegal is a West African with a coverage of local drug manufacturing of arround 15 % of the market.
For the registration of Medicines Senegal follows mainly WAEMU and therefore applies its guidelines. In addition, there are some national texts and specificities of the country to know.
The registration dossier must be in CTD format and in French language.
Drug Registration Authority: The ARP Senegal (Pharmaceutical Regulatory Agency)
The registration of drugs depends on the acceptance of deposit from DPM and on the level of compliance of the file and requires 9 months to 24 months.
AREMA offers you total assistance for your medicines and heath products in Senegal.
.
COUNTRY OVERVIEW: Cote d'Ivoire,, IMPORTANT MARKET AND REGULATORY INFORMATION
Cote d'Ivoire, Abidjan is in West Africa with a population of about 27 million inhabitants, and as an official language French.
Cote d'Ivoire is a member state of the Economic Community of West African States (ECOWAS).
Medicinal products and other health products can only be imported after registration.
An inspection of the manufacturing site of medicinal products is mandatory during the first registrations.
Medicines market in Cote d'Ivoire:
The African market in general is a high-volume import market, in Africa local production accounts for only 3% of world production, while the market is expanding and demand is growing. Cote d'Ivoire imports around 94% of its medicines.
Moreover, introduction of noncommunicable diseases in addition to the major pandemics on the continent, did that the demand for medicines has never been so strong.
Arema offers you the possibility to carry out market studies and field surveys in relation to your products ranges. In addition, AREMA offers you the opportunity to establish the right marketing and distribution way for your products in Cote d'Ivoire.
Registration of medicines:
Cote d'Ivoire is a member of regional institutions, mainly WAEMU and therefore applies its guidelines on the registration of medicines. In addition, there are some national texts and specificities of the country to know.
The registration dossier must be in CTD format and in French language.
The authority governing the registration of medicines is the DPML (Direction of Pharmacy, Medicine and Laboratories) under the supervision of the Ministry of Public Health, whose website: www.dpml.ci
The registration depends on the level of compliance of the file and scheduled commissions, it initially requires to make an appointment for the deposit which will depend on the availability of the authorities then registration will take 12 months to 2 years after the deposit.
.
Country overview: Burkina Fasso , important market and regulatory information
Burkina Fasso is a country in West Africa whose capital is Ouagadougou with a population of approximately 20 million, and as official language French.
Burkina Fasso is a member state of the Economic Community of West African States (ECOWAS).
Medicines can be imported to Burkina Fasso only after registration.
Medicines market in Burkina Fasso:
The African market in general is a high-volume import market, in Africa local production accounts for only 3% of world production, while the market is expanding, and demand is growing. Burkina Fasso has only one manufacturing unit.
Moreover, introduction of noncommunicable diseases in addition to the major pandemics on the continent, did that the demand for medicines has never been so strong.
Registration of medicines:
Burkina Faso is a member of regional institutions, mainly WAEMU and therefore applies its guidelines on the registration of medicines. In addition, there are some national texts and specificities of the country to know.
The registration dossier must be in CTD format and in French language.
Drug Registration Authority: National Agency for Pharmaceutical Regulation of Burkina Faso (ANRP) ​: https://anrp.bf
The registration of drugs depends on the level of compliance of the file and requires 9 months to 18 months.
AREMA offers you total assistance for the registration of your medicines in Burkina Fasso.
We provide regulatory services from the regulatory intelligence, preparation and translation of your dossier, the deposit, follow-up and registration of your various products until the establishment of a system of pharmacovigilance and post-marketing monitoring.
In order to initiate any collaboration or for any consultation you can contact us by email:
• info@arema-international.com
.

New formulation of Carbetocin could save thousands of women’s lives
According to a study published in June 27, 2018, led by WHO in collaboration with MSD for Mothers and Ferring Pharmaceuticals. A new drug formulation of Carbetocin could save thousands of women’s lives
Approximately 70 000 women die every year because of post-partum haemorrhage – increasing the risk that their babies also die within one month.
Currently, WHO recommends oxytocin as the first-choice drug for preventing excessive bleeding after childbirth. Oxytocin, however, must be stored and transported at 2–8 degrees Celsius, which is hard to do, in many countries, depriving many women of access to this lifesaving drug. When they can obtain it, the drug may be less effective because of heat exposure.
The study, published in the New England Journal of Medicine, has shown an alternative drug – heat-stable carbetocin – to be as safe and effective as oxytocin in preventing postpartum haemorrhage. This new formulation of carbetocin does not require refrigeration and retains its efficacy for at least 3 years stored at 30 degrees celsius and 75% relative humidity.
For more information here is the article: https://www.nejm.org/doi/full/10.1056/NEJMoa1805489
and WHO press release : http://www.who.int/news-room/detail/27-06-2018-who-study-shows-drug-could-save-thousands-of-women%E2%80%99s-lives
.
CPP use for Drugs Registration in Africa and Middle east
The Certificate of Pharmaceutical Product (CPP) is a document of the World Health Organization’s (WHO) Certification Scheme on the quality of pharmaceutical products moving in international commerce. CPP also known as CoPP establishes the status of the pharmaceutical product and of the applicant for this certificate in the exporting country. WHO developed CPP in the late 1960s with the aim to provide assurance to its members about the quality of the pharmaceutical products moving in international trade. The CPP is widely required by emerging countries such as African and middle eastern countries in new drugs’ submission processes, post-approval changes and renewal of drugs’ registrations. Some countries in middle east request its legalization by their embassy in country of origin. CPP is more and more replacing marketing authorization in country of origin in these countries and is accurately checked by health authorities as a reference document for drug approval process. For more information : http://www.who.int/medicines/areas/quality_safety/regulation_legislation/certification/guidelines/en/index1.html
.